河南中医药大学第一附属医院(河南省中西医结合儿童医院、河南省中西医结合康复医院)是一所集医疗、教学、科研、预防、保健、康复为一体的三级甲等中医医院,是国家中医临床研究基地、国家中医药国际合作交流基地、国家中医住院医师规范化培训基地。
101、903、G37、G81、G82、G9、100、Y10、Y11、Y21
到工人新村站下车
早上08:00--12:00
下午4:30--17:30(冬季)
15:00--18:00(夏季)
我院周围血管科成功救治一例急性下肢动脉栓塞高龄危重患者
中医药夜市传岐黄 红十字博爱送健康 ——我院中医药夜市热闹开市
岐黄仁术护边关,杏林暖阳戍疆情——我院医疗专家团队圆满完成“送医送药下基层”巡诊任务
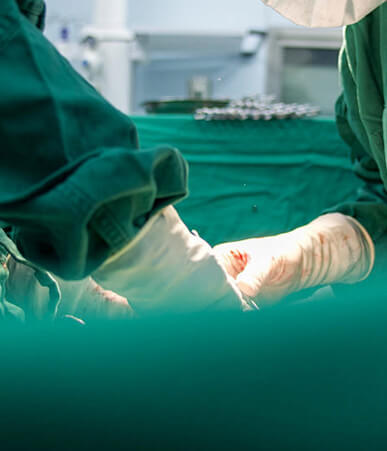
外科手术器械采购项目技术参数公示
河南中医药大学第一附属医院 2025年非中医类别医师学习中医培训招生简章
放射科、磁共振室高端影像设备采购项目技术参数公示
河南中医药大学第一附属医院东院区康复中心诊疗设备采购项目磋商公告
河南中医药大学第一附属医院东院区整形科诊疗设备购置项目公开招标公告
河南中医药大学第一附属医院中药制剂设备购置项目(第四包)- 废标(终止)公告
关于2023年各类各级科技成果奖申报节点的预通知
关于2023年度各类科研项目申报节点的通知
河南省病毒性疾病中医药防治重点实验室开放基金课题申请通知
中心实验室(下设中药药理(呼吸)实验室)举办第六期科研实验技术讲座
我院实验动物房顺利完成《实验动物使用许可证》换证工作
中心实验室简介
河南中医药大学第一附属医院 第一临床医学院(中西医结合学院)儿科医学院 教学简报第44期
第一临床医学院(中西医结合学院)2025年5月24日答辩公告
关于更新伦理审查系统网址的通知
申办方申请伦理审查账号
临床研究伦理审查申请报告指南
【护士节专栏】童心笺语慢递时光——医护亲子互动活动(童真笔绘篇)
【护士节专栏】“光影疗愈”专场,邀你共赴光影之约!
【喜报】以赛促学展风采,以学促用砺精兵——河南中医药大学第一附属医院护理团队在全省心电知识大赛中斩获佳绩
2025年度第一期中医护理适宜技术专项学习录取通知
2025年“心血管病护理及技术专业技能培训”——河南中医药大学第一附属医院基地招生通知
河南中医药大学第一附属医院 中医护理适宜技术专技培训报名须知
2025年5月份进修(护理)录取通知
2025年4月份进修(护理)录取通知
2025年3月份进修(护理)录取通知
传承岐黄,育苗未来——河南中医药大学第一附属医院儿科七区护理团队开展中医药文化进校园系列活动
【喜报】我院肾病科二病区护理团队荣获“第六届河南省健康科普技能大赛”奖项
【护理科普】内分泌一区联合糖尿病护理学组开展科普宣教活动
河南中医药大学第一附属医院2024年招聘工作人员拟聘公示
河南中医药大学第一附属医院2024年护理专业招聘拟聘公示
医院党委副书记董新刚到内科医学部党总支调研指导
医心向党 踔厉奋进:我院举办庆祝中国共产党成立103周年“七一”表彰大会
致敬老党员 “七一”送真情 我院开展走访慰问老党员活动
我院第五届红剧配音大赛圆满落幕,红色之声传递时代力量
三月春“锋”暖,薪火代代传——我院团委开展青年文明号开放周活动
向新而行,岗位建功——我院开展青年文明号开放周活动
2025年6月医德医风事迹公示
2025年5月医德医风事迹公示
2025年4月医德医风事迹公示
地址:河南省郑州市人民路19号
预约挂号电话:0371 - 96129
地址:河南省郑州市中牟县仁信路16号
地址:河南省郑州市博学路与龙子湖南路交叉口东南角
(河南中医药大学龙子湖校区内)